
How does NIPT by GenePlanet work?
The detection of fetal chromosomal abnormalities
In the late 1960s, science and medicine discovered that by analysing the amniotic fluid, they can determine if the fetus has chromosomal abnormalities (such as Down syndrome). Such diagnostic tests are still used today and are called amniocentesis. Amniocentesis is an invasive procedure in which a gynaecologist inserts a needle into the womb under the control of ultrasound. He then takes a small sample of the amniotic fluid. It constains cells from the fetus' skin, and those contain its genetic material. By analysing that genetic material, we can find out if the fetus has any chromosomal defects.
Amniocentesis poses a certain risk, because, in some (usually rare) cases, it may lead to abortion. That is why scientists have been working hard on other, less invasive methods of detecting chromosomal abnormalities, which carry no risk of miscarriage.
NIPT by GenePlanet test
In 2011, NIPT test was introduced into clinical practice. It is an acronym for non-invasive prenatal test. As amniocentesis, it is also based on analysing fetal genetic material, but this time, isolated from pregnant woman's blood. After seven weeks of pregnancy, future mommy's blood contains her fetus' DNA – also called "placental DNA." This kind of testing is non-invasive and thus safe for both the pregnant woman and the fetus.
It is performed by taking a small blood sample from the pregnant woman. The NIPT by GenePlanet test is more than 99% accurate in detecting the most common trisomies present at birth – Down, Edwards, and Patau syndrome.
NIPT by GenePlanet test can also test for other chromosomal abnormalities, including trisomies of other chromosomes, aneuploidies of sex chromosomes, and 60 microdeletion and duplication syndromes. If you would like, it also reveals the gender of the child.
"Placental DNA" in mom's blood
Pregnant woman's blood contains cell-free DNA; short fragments of genetic material which circulate in the blood. During pregnancy, woman's blood contains cell-free DNA fragments of both her and her fetus. To successfully perform the NIPT by GenePlanet analysis, the share of fetal fragments in the blood (fetal fraction) must be 3.5% or more. From week 10 of pregnancy on, fetal fraction is usually high enough to enable NIPT testing.
Our NIPT by GenePlanet methodology
A 10 ml sample of pregnant woman's blood is analysed in our laboratory in Europe. We sequence millions of fragments of fetal and maternal DNA in each sample using reliable, massively parallel sequencing technology.
NIPT by GenePlanet compares the chromosomes in the tested sample with optimal reference chromosomes and accurately determines the presence of genetic abnormalities. If the aneuploidy is present, a small excess or deficiency is detected when counting a particular chromosome.
Some other NIPT tests use the "targeted sequencing" methods, which analyses only the predetermined chromosomes and their parts. In contract, the NIPT by GenePlanet test methodology provides an overview of the fetus' all chromosomes. This enables extremely accurate results (more than 99% accuracy) regardless of the patient's clinical symptoms. It also allows for a more comprehensive range of testing options; trisomies, aneuploidies of sex chromosomes, and microdeletions and duplications.

NIPT by GenePlanet and COVID-19
During the coronavirus (COVID-19) pandemic, pregnant women are wondering if such tests are safe during pregnancy and what is the chance of getting infected with the new coronavirus. Let us reassure you that regardless of the COVID-19 pandemic, the NIPT by GenePlanet test remains safe for you and your baby. If you follow the preventive measures required by the clinic, the risk of getting infected is minimal.
So there's no reason to hesitate.

Most common chromosomal abnormalities
Due to a random error in cell division, one of the parental sex cells may contain 1 chromosome too many, leading to a triple number of chromosomes or trisomy. They have different consequences, but all have a serious impact on the child’s development and health. Trisomies most common at birth are trisomy 21, 18, and 13. The cause of the tripled chromosomes is not yet known, but the consequences of additional chromosomes are well-defined.
Trisomy 21 – Down syndrome
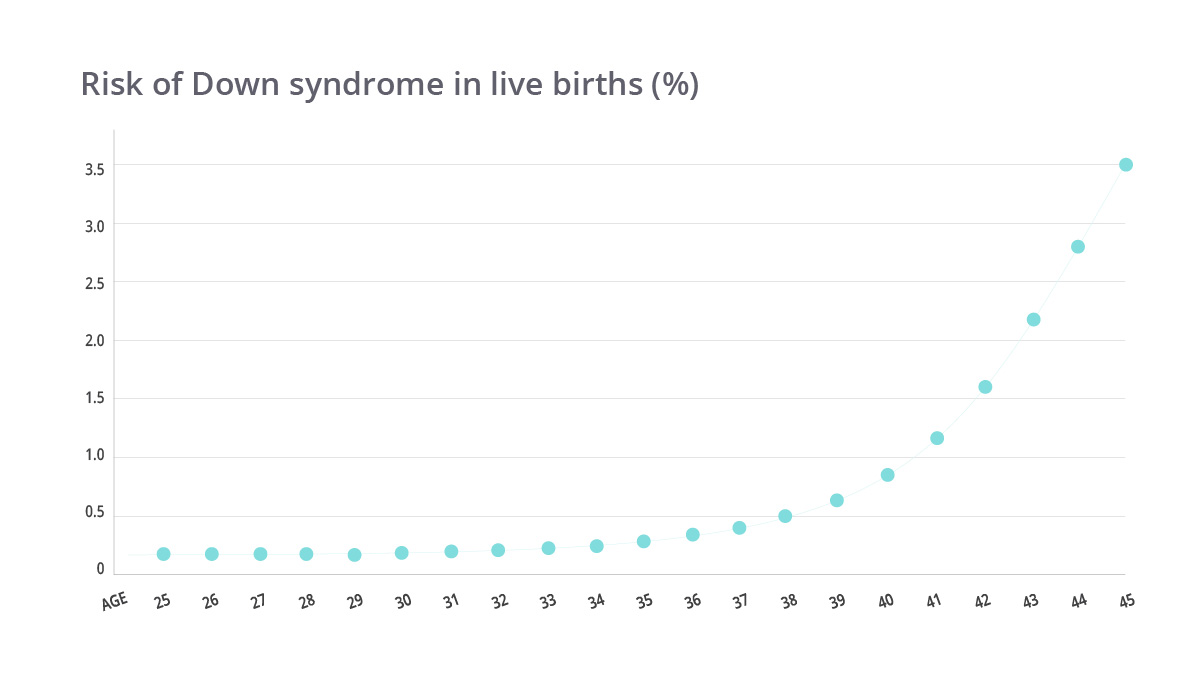
The most common chromosomal abnormality in infants is trisomy of chromosome 21, known as Down syndrome. The risk of the presence of this syndrome increases with the mother’s age. This risk is 1 in 1000 at age 30, while at the age of 35, this risk rises increases to 1 in 400. Unfortunately, 30% of pregnancies with Down syndrome end in miscarriage.
Children with Down syndrome usually have mild to moderate mental development disorders. Early intervention and proper therapy are crucial for ensuring that their life is as normal and independent aa possible. Down syndrome is identified by characteristic facial features, decreased muscle tone (hypotonia), vertical eye folds, common heart diseases, and many other disorders. Only 1% of Down syndrome cases are hereditary.
The consequences of trisomy 21 are mild compared to other trisomies, as people with a mild form of the syndrome can live a relatively normal life.
You can read more about Down syndrome -> HERE
Trisomy 18 – Edwards syndrome
An additional chromosome 18 occurs in about one in 2,500 pregnancies and is three times more common in girls. Edwards syndrome is not usually compatible with life as 80% of such pregnancies end in miscarriage. Less than 10% of babies survive the first year of life.
Abnormalities resulting from Edwards syndrome are visible with ultrasound during pregnancy. At birth, babies exhibit low weight, small jaw, small ears, underdeveloped fingers, and more. Soon, they begin showing mental retardation.
Many children with Edwards syndrome have congenital heart disease and kidney problems. Genetic research shows that most trisomies of chromosome 18 are not heritable.
Trisomy 13 – Patau syndrome
Patau syndrome occurs in about 1 in 5,000 pregnancies. Unfortunately, 97% of such pregnancies end early with miscarriage. However, the vast majority of babies born do not live more than four months. Trisomy 13 is, in most cases, not hereditary.
Patau syndrome often causes abnormal functioning of the heart and brain, back problems, seizures, mental disability, and problems with other organs. At birth, we can spot a cleft lip or palate, undeveloped eyes, clenched fist, extra fingers, and other anomalies.
Each woman can have a child with chromosomal abnormalities. But with age, the risk of some of these abnormalities increases. All women, especially those aged 35 or more, are recommended to perform a prenatal non-invasive test such as NIPT by GenePlanet. As early as week 10, there is enough fetal DNA in the pregnant woman’s blood to enable the detection of potential genetic defects. A safe and simple test, NIPT by GenePlanet tests them with 99% sensitivity.
What to choose: NIPT test or nuchal translucency scan?
Overview of nuchal translucency
Nuchal translucency is the thickness of the pocket of liquid under the skin on the fetus’ back. A gynaecologist can measure it with an ultrasound. The examination of nuchal translucency is both non-invasive and harmless to the fetus. It is usually performed via the abdominal wall, or, less commonly, vaginally.
Nuchal translucency scan primarily assesses the likelihood of chromosomal defects T21 (Down syndrome), T18 (Edwards syndrome), and T13 (Patau syndrome), which are also the most common chromosomal defects of the fetus. These defects are characterised by increased thickness of the pocket of liquid.
Normal nuchal translucency thickness is up to 2.5 mm, and anything above means an increased likelihood of fetal abnormalities. The value indicating a significantly increased chance of chromosomal or heart defect is estimated at 3.5 mm or more. However, the age of the mother is also crucial for the risk assessment itself, as the potential for chromosomal defects increases significantly with the age of the woman.
Gynaecologists perform the nuchal translucency scan between weeks 11 and 14 of pregnancy. The test itself lasts between 15 and 20 minutes. Usually, the gynaecologist performs a thorough general examination of embryonic development and determines the estimated date of delivery. Many gynaecologists also decide to examine the length of the cervix, the cranial anatomy of the fetus, its spine, stomach, kidneys, bladder and determine the presence of all four limbs. With this, the gynaecologist can detect possible other abnormalities in the development of the fetus.
What is the NIPT by GenePlanet test, and how is it performed?
NIPT by GenePlanet is a non-invasive prenatal screening test that detects Down, Edwards, and Patau syndrome, sex chromosome aneuploidies, and deletion and duplication syndromes that can affect fetal development. In cases of deletions and duplications, the number of chromosomes is normal, but only a fraction of the chromosome is missing or duplicated.
![]()
Only 10 ml of mother’s blood is required to perform the NIPT test, as cell-free fragments of fetal DNA are present in maternal blood. These are short fragments of DNA circulating in the blood. And by taking a sample of the mother’s blood, we can obtain the genetic material of the fetus and thoroughly examine it using NIPT by GenePlanet technology.
This method offers extremely accurate results (more than 99% accuracy for the three most common trisomies) regardless of the patient’s clinical symptoms. The test also offers a more comprehensive range of testing options, including other trisomies, sex chromosome aneuploidies, and deletion and duplication syndromes.
The test can be done from week 10 of pregnancy when the mother’s blood contains enough fetal hereditary material. This means that the fraction of fetus’ cell-free DNA has reached the limit of 3.5%, which is the lowest amount for performing the NIPT by GenePlanet test. As it only requires a sample of pregnant woman’s venous blood, the NIPT test is entirely risk-free for her and the fetus. The sample goes to the lab, and the results can be available within 6-8 working days.
Accuracy of results
Overview of nuchal translucency
The biggest drawback of the nuchal translucency scan is that it identifies only between 70 and 80% of fetuses with Down syndrome. Out of 100 embryos examined, the presence of the syndrome won’t be identified in 20 to 30 cases. Therefore gynaecologists usually suggest adding a double hormonal test (measuring betaHCG and PAPP in the mother’s blood), which increases the positive predictive value to 85-90%.
A nuchal translucency scan also provides approximately 5% of false positives, which is problematic as this may unnecessarily expose pregnant women to the risk of invasive diagnostic tests.
![]()
Risk of 1:300 or more?
If a gynaecologist concludes that the risk of chromosomal abnormality in the fetus is 1:300 or higher, he will suggest further tests. The only way to detect chromosomal abnormalities with 100% certainty is an invasive diagnostic test – chorionic villus sampling or amniocentesis. Such tests have up to 1-2% risk of abortion, so it is not an easy decision.
Fortunately, you can choose to take the NIPT by GenePlanet test before the invasive test.
The NIPT by GenePlanet test
The NIPT by GenePlanet test is much more accurate than the nuchal translucency scan. Its detection rate for the three most common trisomies present at birth is higher than 99%.
A significantly lower percentage of false-positive results is also a significant advantage:
- for Down syndrome (T21) only 0.05%,
- for the most common trisomies, 0.14% overall.
As a result, fewer pregnant women are unnecessarily exposed to the risk of invasive diagnostic methods.
![]()
Which test to choose?
The nuchal translucency scan and the NIPT test are not mutually exclusive. The nuchal translucency scan can also detect other morphological abnormalities which a genetic test cannot. At the same time, the nuchal scan is not as accurate as NIPT by GenePlanet. To have a truly peaceful pregnancy, choose to do both.

Is chorionic villus sampling dangerous?
The placenta is the organ surrounding the developing fetus, providing oxygen and nutrients, and removing waste products. The chorionic villi are wispy projections of placental tissue with the same genetic makeup as the fetus. To analyse that genetic material, doctors take a sample of chorionic villus tissue.
The chorionic villus sampling is performed between weeks 11 and 13 of pregnancy, most often in week 12. Gynaecologists recommend it to women with an increased risk of their child having chromosomal abnormalities and/or monogenic diseases. This risk is usually higher in older pregnant women (after 37) as well as when other first-trimester screening tests (nuchal scan, double or quad marker scan) show an increased risk of a particular genetic abnormality. Chorionic villus sampling is also advised to couples with abnormal results of a non-invasive prenatal test (NIPT by GenePlanet), which detects the most common chromosomal abnormalities with 99% accuracy.
How is chorionic villus sampling performed?
The process involves taking a small sample of tissue from the developing placenta. With the help of an ultrasound, the doctor determines the position of the fetus and placenta and then penetrates the abdominal and uterine wall with a thin needle. He then removes a sample of the chorion villus tissue. This tissue is then sent to the lab, where they analyse the child's DNA and determine if any of the tested genetic abnormalities are present.

Very reliable results on the one hand and the risk on the other
The primary purpose of the chorionic villus sampling is to detect Down syndrome and other chromosomal and genetic abnormalities. It detects three most common changes in the number of chromosomes (trisomy 21, 18, and 13), i.e. Down, Edwards, and Patau syndrome, as well as changes in chromosome structure (duplication, deletion, and translocation). The positive side of the chorionic villus sampling is its very high – more than 99% – diagnostic accuracy. But because of its invasiveness, it also has drawbacks: the test can lead to complications such as unwanted miscarriage, which happens in 1–2% of cases.

However, if a woman receives a high-risk result from a screening test, it should be confirmed with a diagnostic method, such as chorionic villus sampling or amniocentesis.

Down syndrome
We remember from basic biology lessons that people have 23 pairs of chromosomes present in each body cell, a total of 46 chromosomes. We received 23 from our mother and 23 from our father. Each of those chromosomes carries a complex genetic record that determines our traits and functioning of our bodies. However, people with Down syndrome have an additional copy of chromosome 21. Because they carry tree chromosomes 21, this condition is also called trisomy 21.
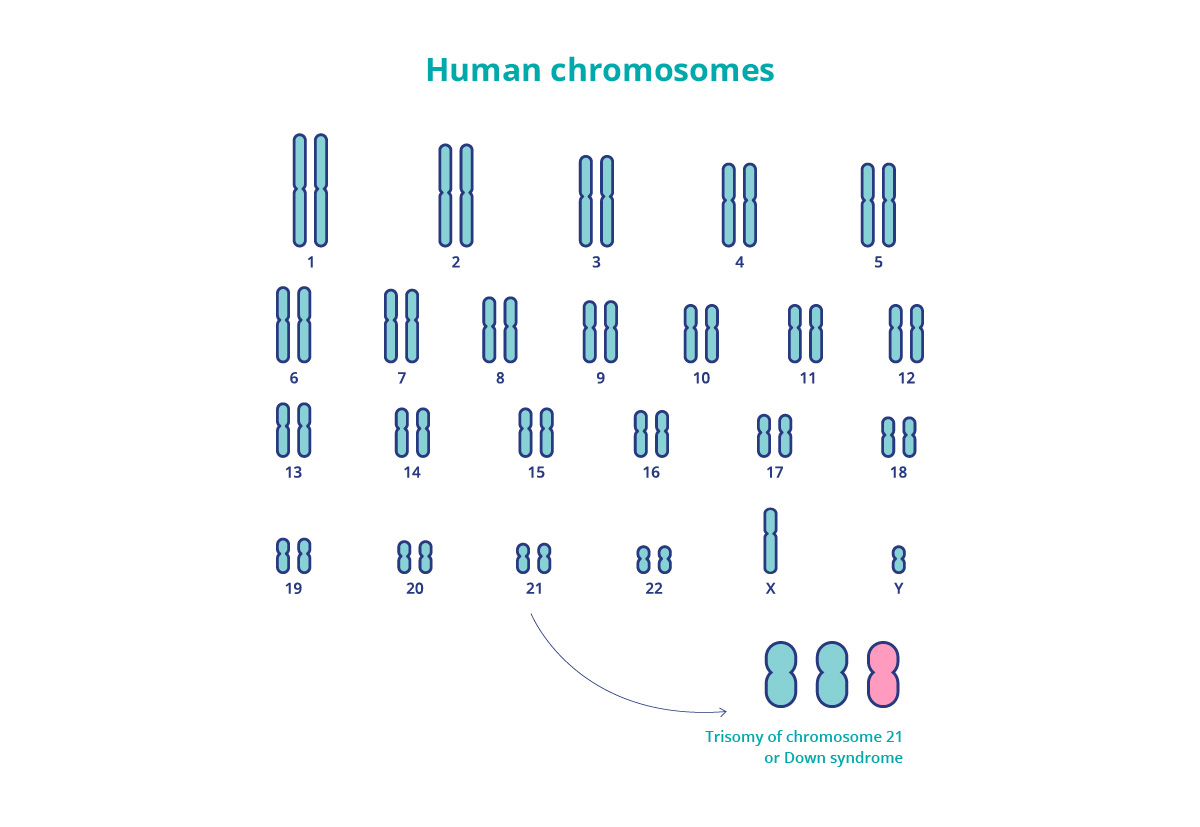
Down syndrome
“Syndrome” means a set of physical and mental symptoms (or characteristics) that continuously appear together, in this case, in the presence of a third copy of chromosome 21. The name “Down” comes from doctor Langdon Down, who first described these characteristics. The additional chromosome that causes Down syndrome was discovered in 1959. The development of genetics helped us understand trisomies much better.
Most common chromosomal abnormalities
The frequency of Down syndrome is 1 in 700
People with Down syndrome usually have distinctive facial features, low muscle tone, lower stature, and stocky physique. Their mental and physical development is slower. But these common characteristics do not mean that all people with Down syndrome are the same and will have equal development. Down syndrome varies in its intensity. About half of babies with Down syndrome experience heart defects, but modern medicine can treat them.
We know much more about Down syndrome today than we used to, which makes it easier for us to understand and live with this condition. As a result, the life span of people with the syndrome has changed significantly. Back in 1982, the average life expectancy of people with Down syndrome was only 25 years, mostly spent in psychiatric institutions. Today, people with this syndrome live on average for 60 years, and their lives are much fuller.
What causes Down syndrome?
Trisomy 21 happens because to an error in cell division during meiosis. The causes of it are still unclear to this day and can occur in any pregnancy. But the fact is that the chance of Down syndrome increases with the age of the pregnant woman.
For example, the risk of having an additional copy of the 21st chromosome is 1 in 1,150 when women are 21 years old. While for 36-year-old women, the risk increases in 1 in 210 and women at 42 have 1 in 40 chance. There is also a 30% chance of miscarriage if you are carrying a child with this kind of genetics mistake.
Is It possible to detect Down syndrome before the baby is born?
Yes, trisomy 21 can be detected before birth. Because today we have a lot of information available on the syndrome it is important to identify its presence as soon as possible. The unexpected birth of a child with Down syndrome is a massive shock to the parents. Fortunately, Down syndrome can be detected today with prenatal tests that analyse potential presence of chromosomal abnormalities.
We can discover Down syndrome in the fetus with non-invasive prenatal tests, such as nuchal translucency scan and NIPT test. As early as from week ten of pregnancy, the NIPT by GenePlanet test detects this type of genetic defect with 99% accuracy. For final confirmation, however, gynaecologists and physicians use invasive methods like amniocentesis and chorionic villus sampling.
What to choose: NIPT test or nuchal translucency scan?
The discovery of Down syndrome at an early stage of pregnancy allows parents to learn more about the syndrome and talk to doctors, experts, and parents who have children with Down syndrome. This way, they can create a realistic picture of living with the syndrome, gather knowledge, and prepare for the future even before the baby’s born.
Living with Down syndrome
Each person has their character, talents, and abilities. This also applies to people with Down syndrome. Although children with trisomy 21 have some common characteristics, they are very distinctive and unique.
In the past, people with the syndrome have been underestimated and misunderstood. It has led to strong prejudice, portraying them as entirely incompetent, helpless, and condemned to life in institutions. That is, of course, not the case. Today they live significantly better than they used to, as society provides parents with much more support, therapies, and information about appropriate care and education.
Most children with Down syndrome learn to walk and talk. Most know the basics of reading and computation. Some go to mainstream elementary schools; others attend tailored programmes. They have friends, hobbies, and later even jobs. Nowadays, in more advanced societies, people with Down syndrome also have more opportunities to work and can grow into semi-independent individuals. Some even get married and get a driver’s license.
Emotional intelligence
If people with a “normal” chromosome count often feel superior compared to people with Down syndrome, at least in one area they should not: emotional intelligence. People with trisomy 21 have much more developed emotional intelligence. They have, in the absence of perceptual and speech abilities, emotional experience in abundance. They are deeply compassionate, eager to help, and kind.
Many parents will tell you that a child with Down syndrome has changed their life’s outlook and taught them a lot of positive things. Some say that the extra chromosome may slow the development, but it makes it up for with love and joy.
But that doesn’t change the fact that discovering Down syndrome in a child is very stressful for parents. They can experience a range of emotions and can feel scared and lost. At such times, parents can turn to institutions for help, consult doctors, or contact parents who have personal experience. The realisation that they are not alone in this is of paramount importance.

What is amniocentesis?
Amniocentesis is most commonly performed between weeks 16 and 18 of pregnancy
Although the amniotic fluid is already present earlier, we have to wait for the amnion to fuse with the outer chorion. This usually happens in week 15 of pregnancy. Therefore, amniocentesis is performed from the 15th week of pregnancy onwards, most often until weeks 18 or 20. In the second trimester of pregnancy, this procedure can detect fetal chromosomal abnormalities and mutations in genes.
Doctors sometimes recommend amniocentesis even later, if the gestational sac leaks and they want to determine whether the uterus may be infected and needs to be treated. Amniocentesis can also help identify anaemia in infants whose mothers are RhD negative and might need a blood transfusion. In some cases, doctors decide for amniocentesis just before the scheduled date of delivery, as the procedure can help assess the maturity of the child’s lungs and predict whether he can breathe on his own.
The risk of chromosomal defects increases with age
Although amniocentesis is performed for a variety of reasons, the main is an increased risk of a chromosomal defect. This risk varies in each pregnant woman and generally increases with age, especially in pregnant women over the age of 37. From the age of 30, the risk of having a child with Down syndrome triples every three years.

The results are 99.9% reliable
Amniocentesis’ advantage is its extremely high predictive accuracy. It is 99.9% accurate in determining whether the fetus has chromosomal abnormalities, neural tube defects, or genetic defects. Unfortunately, it cannot predict the extent of these defects. Ultrasound and measurement of alpha-fetoprotein levels can help assess fetal defects.
Is amniocentesis dangerous for my child and me?
It is a usually safe, often urgent procedure. But as all invasive procedures during pregnancy, it poses certain risks. Because the doctor enters the womb with a needle, amniocentesis can trigger miscarriage and loss of the fetus. The chance of such a complication is, on average, 1-2 per cent.
Because the method is invasive and the needle comes near the infant, in rare cases it may damage both the fetus and the mother. Infection and other complications may also occur, but the likelihood is extremely low.

Should all pregnant women undergo amniocentesis?
Definitely not. The high risk of Down syndrome is first assessed by gynaecologists using conventional screening methods such as nuchal screening, double and quadruple marker testing, or non-invasive prenatal screening test or NIPT by GenePlanet.
Amniocentesis is precise, but because of the undesirable effects of invasive methods, doctors recommend that pregnant women with increased risk of chromosomal defects of the fetus should first do a non-invasive risk assessment methods. Only later, with unfavourable results, should they resort to invasive procedures such as amniocentesis.


I’m pregnant, what now?
Can the test be wrong?
We know blood and urine-based pregnancy tests. If you took the test at home, you probably chose the urine test. Two bars appear on it if the human chorionic gonadotropin (hCG) or the “pregnancy hormone” is present in the body. The level of this hormone varies from day to day and also changes throughout the day (depending on how much a woman drinks and how concentrated her urine is). That is why it is recommended to use the first morning urine for the test. Home pregnancy tests are, of course, not 100% reliable. Sometimes they’re negative, even though a woman is pregnant. This can happen because the level of the pregnancy hormone is still too low to be detected by the test. The hCG hormone level might also be so high (in later pregnancy) that the test also shows a negative result, even when pregnancy is evident. The urine test can also be falsely positive, which often happens after miscarriage or ectopic pregnancy, as the body needs time to normalise the level of the pregnancy hormone.
If the test showed you a positive result ad your period is late, you are most likely pregnant! You can, of course, repeat the test, use another brand to make sure, but probably there is no need for a double-check.
What now?
Have breakfast first. Probably, you did the test early in the morning, and now you’re running through the fridge – what can I even eat during pregnancy? Well, you can probably forget gorgonzola for a while, but keep in mind that pregnancy is not a disease. However, it is a special condition in which, for the sake of health, you have to give up some items on your menu. Those include raw or uncooked foods such as eggs (or dishes prepared from them) meat and fish (beef tartare, Prosciutto, sushi, smoked salmon), unpasteurised milk and milk products, soft and mouldy cheeses. If your hand now reached into a drawer of cans, beware! Don’t exaggerate with cans of tuna and pâtés, as they contain a lot of mercury and vitamin A, which can be harmful to the fetus in large quantities. And now you know what a healthy pregnancy breakfast can look like. Think toast with eggs, fruits, nuts, yoghurt. Above all, listen to yourself and your body!

Then call the gynaecologist to schedule for your first examination and confirmation of the pregnancy. And don’t be surprised that you probably won’t get an appointment this afternoon. If you don’t have any problems, the first gynaecological exam is recommended between weeks 8 and 12. Why? Because before that time, the gynaecologist can’t confirm the pregnancy with an ultrasound. The first pregnancy check-up is very emotional, so prepare for the tears of happiness. A gynaecologist uses vaginal ultrasound for examination, inspecting the uterus and fetus. When confirming a pregnancy, they’ll ask about the first day of your last period, so have this information ready. Based on it, the doctor can calculate the approximate date of birth. Urine and blood sampling will follow, sometimes a test for toxoplasmosis. Then the doctor will talk to you and clear up all your dilemmas.
Is my child healthy?
Waiting for the first pregnancy check-up often feels like an eternity, so pregnant women are left with many unanswered questions. They can cause worry, fear, discomfort, or anxiety. But future mom – take a deep breath and believe that your child is healthy. You can confirm it quickly and easily with NIPT by GenePlanet, a non-invasive prenatal test. From week 10 of pregnancy, it can accurately detect if the child has any genetic abnormalities. The test poses no risk to you or the baby, but it does ensure that you have a peaceful and cheerful pregnancy.

Should I already feel that I’m pregnant?
You must have automatically started caressing your tummy and taking care not to lie down on your stomach so you don’t “squash” the baby. Don’t worry about that; you can still sleep on your stomach for the first few months. Later it’s going to become more challenging because your stomach will be in the way and you’ll probably be “doomed” to sleep on your left side until you give birth.
The first signs of pregnancy are not kicks in the abdomen, but you can feel cramps and menstrual-like pain in the uterus area. Many pregnant women sleep a lot in the first weeks. They are tired, which is normal – your body is creating something extraordinary. It’s building an environment where a child will evolve. Your body is fantastic, be aware of it and give it a rest if it needs it. The pregnancy hormone also sends you to the bathroom very often; more frequent peeing is one of the first signs of pregnancy. The hormone hCG speeds up blood flow through the kidneys and helps the body get rid of excess fluid. At the same time, your uterus expands, creating space by retreating and pressing on other organs, including the bladder.

You’ve undoubtedly heard of pregnancy sickness. Science has not yet fully discovered why it happens, but pregnancy hormones, especially progesterone, and genetics are thought to play a significant role. Pregnancy sickness accompanies most women’s first months of pregnancy and then slowly subsides. Your smell will be sharper, more sensitive. You may remove all the onions from your home because you’ll be so sick you won’t be able to smell them. Almost all pregnant women also experience a strong desire for certain foods and a strong aversion to others. Know that all this is normal and that it is your hormones that are “out of control”.
How and when should I tell I’m pregnant?
If you haven’t already, share the news with your partner first. You could do it by simply saying: “You will be a daddy!” or you can make it more exciting and prepare a surprise he will never forget. If you’re home alone at the moment, and if you have time, maybe think about how you’re going to tell him the happy news. You can also record it secretly to create a memory that you can relive over and over again. Then decide together when you’re going to tell the world. Some do so immediately, others wait for the results of the NIPT by GenePlanet test, and some couples tell the happy news after three months of pregnancy when the chance of miscarriage is significantly lower. The decision, of course, is all yours.
When should I buy a stroller?
The answer is simple: not now. First, take care of other, more important things. First on your list is a healthy diet; make sure you’re getting enough folic acid, iron, and other necessary nutrients. Be regularly active outside in the fresh air. Also, make sure your pregnancy is calm. If you’re worried about your baby’s health, do a NIPT by GenePlanet test, take a deep breath, and dive into the pregnancy. Take care of your body. Use gentle soaps and shower gels because the skin is often sensitive during pregnancy. Use a cream or oil against stretch marks on your stomach and breasts every night to make sure your skin stays elastic and nurtured.
And then comes happy shopping; car seats, clothes, accessories for the baby’s room, stroller, and more. And while spending, keep in mind that what the child needs most is love and a safe nest that only you and your partner can provide. Even the most expensive and softest blanket can’t compete with mom’s lap.


Sex chromosome aneuploidies
They can occur in both men and women, but not all in both sexes. Sexual chromosome defects cause various clinical signs from physical characteristics, developmental and learning disorders to infertility.
Sex chromosome aneuplodies among men
Klinefelter syndrome (XXY)
Men affected by Klinefelter syndrome have an additional X chromosome in each body cell.
The main symptom is small testicles, not producing enough testosterone before birth and during puberty. Testosterone deficiency, however, inhibits the normal development of male characteristics. Men affected by Klinefelter syndrome have a less hairy face and body. They may also develop breast tissue, increasing their risk of breast cancer; but this risk is still lower than in women. In many cases, the syndrome leads to infertility, and it can lead to weaker muscles, higher stature, and inadequate libido. Usually, the syndrome does not affect intelligence, but reading and speaking problems may occur in some cases. There is no cure for the syndrome, but various physical, speech, and hormonal or testosterone therapies can help. From time to time, the adjustment of learning methods is also necessary.
Klinefelter syndrome occurs randomly and is not hereditary. It is present in 1 to 2 live-born boys per 1,000 births. This makes it the most common among all syndromes caused by sex chromosome abnormalities. It is also more common than Down syndrome. The reason is most likely the fact that life with this syndrome can be reasonably normal. As adults, most men with the syndrome lead lives similar to those without it.

Jacob's syndrome (XYY)
XYY syndrome, also called Jacob's syndrome, affects men and is caused by an additional Y chromosome.

Affected individuals are usually very tall, and many have severe acne problems at a young age. Some also experience learning difficulties, behavioural issues, and impulsivity. However, the presence of an additional Y chromosome does not lead to infertility and a decrease in intelligence, so people with this syndrome can live a normal life.
Sex chromosome aneuplodies among women
Turner syndrome (X)
Turner syndrome is caused by a total or partial absence of sex chromosome X in women.
The syndrome has a wide range of symptoms: the two most common are low stature and underdeveloped ovaries. The latter results in the absence of period, infertility, and less developed breasts. Women with only one X chromosome may also experience heart defects, diabetes, and thyroid problems. However, most do not have problems with mental development and are normally intelligent.

Some women may have visual, hearing, or spatial performance problems. Turner syndrome is often incompatible with life, and the affected fetus does not survive until birth. If born, there is no cure for Turner syndrome, but therapies can help alleviate symptoms. Growth hormone injections in childhood can help with more normal growth, and estrogenic treatments can help develop breast and ovarian tissue.
Turner syndrome is not usually hereditary. It occurs during the formation of the parent's reproductive cells or during cell division at an early development stage. Turner syndrome appears in about one girl among 2,000 to 5,000 births. Chromosome abnormality is sometimes present in only some of the cells – in this case, the abnormality is called Turner syndrome with mosaicism. In such cases, a person usually has fewer or no symptoms at all.
World-famous actress Linda Hunt is one of the most successful and recognisable actresses despite being diagnosed with Turner Syndrome.

Source: s_bukley/Shutterstock.com
Triple X syndrome (XXX)
Triple X syndrome occurs when a woman has an additional X chromosome in each cell.

Some women have very mild symptoms or none at all, so the syndrome might even remain undiagnosed. Women with this syndrome are often taller than average and may have difficulty learning. Delayed speech development and underdeveloped motor skills (such as sitting and walking) can also occur. They can have weak muscle tone (hypotonia), behavioural disorders and emotions difficulties. About 10 per cent of those affected with Triple X syndrome experience kidney problems. Some of these problems can be alleviated or eliminated with speech and physical therapies. Women who have an additional X chromosome present have no fertility problems and usually have normally developed intelligence.
The syndrome appears in about one in 1,000 live births. It is estimated that as many as 90 per cents of girls with the syndrome are left undiagnosed as they have zero or very few symptoms, and can live a full and normal life.
Although sex chromosome defects tend to have milder consequences than other chromosomal abnormalities, it is still crucial to know about them. This way, parents can properly prepare for the birth of a child with the syndrome. The NIPT test can predict the presence of sexual chromosome defects from the 10th week of pregnancy. If the analysis detects such aneuploidies, the parents have enough time to learn about the health consequences and provide the child with appropriate post-birth assistance and/or therapies.

Deletion and duplication syndromes
Deletion
A chromosomal deletion occurs when a fragment of the chromosome is lost. It can happen on any chromosome and can be of different size. The result of chromosomal deletion is the loss of genetic material, which normally provides instructions for the body. The consequences of the deletion depend on the size of the lost part and its location on the chromosome: in other words, what information did this missing fragment of genetic matter contain.
Duplication
Chromosomal duplication is the opposite of deletion: it is the doubling of a part of the chromosome. As a result, the body has too much genetic material or (instructions.). The consequences of a duplication also depend on the size and location of the duplicated part of the chromosome.

Clinical characteristics of deletions and duplications may include developmental retardation and intellectual developmental disorders, slowed growth, behavioural disorders, feeding problems, low muscle tone, seizures, characteristic facial features, and other abnormalities. Clinical characteristics vary significantly between deletions and duplications, so if chromosomal abnormalities are detected, we recommend consulting an expert who will be able to answer your questions.
92 syndromes of deletion and duplication (NIPT Pro)
1p36 Deletion Syndrome
1p36 deletion syndrome is characterized by characteristic craniofacial features, intellectual disability, seizures, skeletal abnormalities, and brain and cardiac defects. Prenatal ultrasound may identify certain structural anomalies such as cardiac defects, agenesis of corpus callosum, hydrocephalus, and microcephaly. However, a normal ultrasound does not rule out the underlying anomaly. Lifespan is variable but can be normal.
1q41–q42 deletion syndrome
Key features of this syndrome include a significant developmental delay and distinct facial dysmorphisms such as deep-seated eyes, broad nasal tip, and depressed nasal bridge. Additionally, some cases may exhibit conditions such as abnormally small head size (microcephaly), a gap in the roof of the mouth (cleft palate), clubfeet, seizures, and short stature.
1p32–p31 deletion syndrome
Key features of this syndrome include developmental delay, issues with the structure of the brain (like a missing or underdeveloped part called the corpus callosum), and certain facial features such as a larger head (because of extra fluid in the brain), ears that are lower down, nostrils that face forward, and a small jaw. Problems with the urinary tract, like a backward flow of urine and difficulty controlling it, are also common. Other clinical features may include weak muscle tone (hypotonia), tethered spinal cord, Chiari type I malformation, and seizures.
2p16.1–p15 deletion syndrome
Key features of the 2p16.1–p15 deletion syndrome include delayed psychomotor development, intellectual disability, and variable but distinctive dysmorphic features, including a small head, receding forehead, an increased distance between the inner corners of the eyelids, a fold of skin covering the inner corner of the eyes, eyes that are short and slant downward, droopy eyelids, a wide and high nose, a jaw that is set back, a flat area between the nose and upper lip, a small mouth with a high and narrow roof, and a lower lip that turns outward. Patients might also show certain behaviors, including features similar to autism, and have differences in the structure of their brains.
2q33.1 deletion syndrome (Glass syndrome)
Key features of the Glass syndrome include an intellectual disability of variable severity and dysmorphic facial features, including an abnormally small lower jaw (micrognathia), downward slanting palpebral fissures, a gap in the roof of the mouth (cleft palate), and crowded teeth.
2q31.1 duplication syndrome
2q31.1 duplication syndrome, also called mesomelic dysplasia, , is a rare skeletal genetic syndrome. Key features include symmetric shortening of the middle segments of limbs, making a person shorter than usual, rapid eye movements (nystagmus), and hand issues like a thumb with three parts, skin joining fingers, usually between fingers 3-4, and some individuals might have clubfeet.
2q31.1 microdeletion syndrome
This syndrome is characterized by moderate to severe developmental delay, short stature, facial dysmorphism, and variable limb defects.
2q37 deletion syndrome
2q37 deletion syndrome presents with mild to moderate developmental delay or intellectual disability, shortened fingers, short stature, obesity, weak muscle tone (hypotonia), specific facial features, abnormal behavior, autism spectrum disorder, joint hypermobility or dislocation, and scoliosis.
2q duplication
2q duplication means that a part of the long arm of chromosome 2 is duplicated. Key features of this duplication include moderate psychomotor delay, mild intellectual disability, facial dysmorphism (high hairline, prominent forehead, widely spaced eyes (hypertelorism), upward-slanting eye openings, large, low-set and/or posteriorly rotated ears, depressed/broad nasal bridge, prominent nasal tip, and thin upper lip. Additionally, individuals with this condition may exhibit finger abnormalities and either normal or above-average body measurements. Genital anomalies and short stature may be observed on occasion.
3pter-p25 deletion syndrome
Key features of this syndrome include low birth weight, psychomotor and growth retardation, a smaller head, and specific facial features like droopy eyelids and a small jaw. Clinical features might also include extra fingers or toes, renal anomalies, a gap in the roof of the mouth (cleft palate), congenital heart defects, and gastrointestinal anomalies. Although patients typically have severe to profound intellectual disability, rare patients with a 3p26-p25 deletion have normal intelligence or only mild abnormalities.
Dandy-Walker syndrome (DWS)
Dandy-Walker syndrome is a rare genetic syndrome caused by a deletion in chromosome region 3q22-q24. Key features of this syndrome include affected brain development, specifically the part called the cerebellum, which is responsible for coordinating movement. The syndrome’s main feature is the enlarged fourth ventricle in the brain. Up to half of the affected individuals have intellectual disability, delay in the development of motor skills, muscle stiffness, and seizures. The incidence is approximately 1 in 25,000-35,000.
3q13.31 deletion syndrome
Key features of this syndrome include major developmental delay (including speech delay), postnatal overgrowth, characteristic facial features with a prominent or broad forehead, high arched palate, a short space between the nose and upper lip, and protruding lips. Brain and central nervous system anomalies, including the absence or incomplete development of the corpus callosum (tissue that connects the left and right sides of the brain), and abnormal male genitalia can also occur.
Distal chromosome 3p duplication
Distal chromosome 3p duplication means that a part of the distal region of the short arm of chromosome 3 is duplicated. Key features of this duplication include specific craniofacial dysmorphism associated with psychomotor delay, moderate to severe intellectual disability, cardiac and urogenital abnormalities, as well as seizures and presence of whorls on fingers.
3q duplication
3q duplication means that a part of the long arm of chromosome 3 is duplicated. The severity of the condition and the signs and symptoms depend on the size and location of the duplication and which genes are involved. Key features of this duplication include distinctive facial features, hirsutism (excessive hair growth in women), small head size (microcephaly), intellectual disability, slowed growth, and abnormalities of the hands, feet, genitourinary system, kidneys, and/or heart. Various other neurologic abnormalities or birth defects affecting other parts of the body may also occur. About one third of babies with chromosome 3q duplication do not survive past the first year of life, often due to heart defects or infections.
4p16.3 deletion syndrome (Wolf-Hirschhorn syndrome)
Wolf-Hirschhorn syndrome is a rare genetic syndrome characterized by birth defects, intellectual disability, and other serious medical issues. Key features of this syndrome include prenatal-onset growth deficiency followed by postnatal growth retardation and hypotonia with muscle underdevelopment, typical craniofacial features in infancy consisting of the characteristic appearance of the nose, microcephaly, intellectual disability of variable degree, seizures, skeletal abnormalities, congenital heart defects, hearing loss (mostly conductive), urinary tract malformations, and structural brain abnormalities.
4q21 deletion syndrome
Key features of this syndrome include progressive growth restriction, psychomotor retardation, and distinct facial features like a wide forehead, short space between the nose and mouth, widely spaced eyes (hypertelorism) and downturned corners of the mouth. People with this syndrome also have significant intellectual disabilities and absent or severely delayed speech.
4p duplication
4p duplication means that a part of the long arm of chromosome 4 is duplicated. The severity of the condition and the signs and symptoms depend on the size and location of the duplication and which genes are involved. Key features of this duplication include developmental delay, intellectual disability, behavioral problems, and distinctive facial features (smaller head, increased distance between eyebrows and eyes, enlarged ears, bulbous nose with flat or depressed nasal bridge, a long space between the nose and upper lip, lower jaw that is set back with a pointed chin). Additional features include skeletal (rocker bottom feet, arachnodactyly, camptodactyly) and renal malformations, cardiac defects, ocular abnormalities, and abnormal genitalia in males.
Distal chromosome 4q duplication
Key features of this duplication include growth deficiency, mental retardation, distinctive malformations of the skull and facial (craniofacial) region, including an unusually small head (microcephaly), malformed ears, and a prominent nasal bridge, and/or defects of the hands and feet. In some cases, additional physical abnormalities may also be present, such as structural defects of the heart that are present at birth (congenital heart defects), genital abnormalities in affected males, urinary tract defects, and/or other findings.
Distal chromosome 4q deletion
Key characteristics of this deletion include a variable combination of craniofacial, developmental, digital, skeletal, and cardiac features: weak muscle tone (hypotonia), developmental delay, growth deficiency, a gap in the roof of the mouth (cleft palate), cardiovascular malformations, abnormalities of the hands and feet and typical dysmorphic features, such as abnormally small head size (microcephaly), rounded facies, small eyes, broad nasal bridge, upturned nose, full cheeks, small mouth and chin.
Cri-du-chat syndrome (5p deletion syndrome)
5p deletion syndrome, also known as Cri-du-chat syndrome, is a genetic syndrome characterized by birth defects, intellectual disability, and other serious medical issues. Key features of this syndrome include significant intellectual disability, speech delay, cat-like cry, dysmorphic features, cardiac defects, and microcephaly. There is a 10% mortality rate in the first year. Approximately 1 in 20,000 to 1 in 50,000 live births have this condition.
5q14.3 deletion syndrome
Key features of this syndrome include severe intellectual disability, absent speech, stereotypic movements, and epilepsy. Unusual facial features include a high broad forehead with a variable small chin, short nose with anteverted nares (nostrils that open to the front rather than downward), large open mouth, up-slanted palpebral fissures (outside corners of the eyes that point downward), and prominent eyebrows.
5q12 deletion syndrome
Key features of this syndrome include delayed psychomotor development, intellectual disability, speech delay, decreased body mass index, long arms, fingers, and toes, and facial dysmorphism (large forehead, prominent nose, long space between the nose and upper lip, and small chin).
5p13 duplication syndrome
Key features of this syndrome include global developmental delay, intellectual disability, autistic behavior, weak muscle tone (hypotonia), a larger head and distinct facial features (a prominent forehead, short eyes, and small or unusual ears). Other associated clinical features include sleep disturbances, seizures, aplasia/hypoplasia of the corpus callosum (developmental abnormalities affecting the bundle of nerve fibers that connects the left and right hemispheres of the brain), and skeletal abnormalities (large hands and feet, long fingers and toes).
5p duplication
Key features of this duplication vary widely but always include severe intellectual deficit. Other clinical features may include facial dysmorphism, growth abnormalities, neurological problems, heart defects and other organ abnormalities.
6pter-p24 deletion syndrome
Key features of this syndrome include developmental delay, delayed motor development, intellectual disability, weak muscle tone (hypotonia), and abnormal skull shape. Common clinical features include congenital heart defects, structural eye defects, widely spaced eyes (hypertelorism), anterior eye chamber abnormalities, palatal and dental abnormalities, abnormalities in nerve cells or their connections, and anomalies of the arms, legs, hands, and feet.
6q24-q25 deletion syndrome
Key features of this syndrome include developmental delay, intrauterine and postnatal growth restriction, facial dysmorphism (midface hypoplasia, widely spaced eyes (hypertelorism), broad bridge of the nose and backwards rotated ears), heart abnormalities, delayed bone age, small hands and feet, and hearing loss.
6q11-q14 deletion syndrome
Key features of this syndrome include weak muscle tone (hypotonia), psychomotor delay, short stature, skeletal/limb anomalies, umbilical hernia, and urinary tract anomalies, as well as characteristic facial features including upward-slanting eyes, high-arched palate, and low-set and/or dysplastic ears.
6p deletion
6p deletion means that a part of the short arm of chromosome 6 is missing. The severity of the condition and the signs and symptoms depend on the size and location of the deletion and which genes are involved. Key features of this deletion include intellectual deficit, developmental delay, eye abnormalities, hearing loss, hand anomalies, widely spaced eyes (hypertelorism), and heart defects.
6q15-q23 deletion syndrome
Key features of this syndrome include small or very small babies, often delivered by Caesarean section. Infants with this syndrome may exhibit reluctance or inability to feed, abnormal muscle tone (either floppiness or tightness), and typically require special care. Additionally, they may have heart problems, minor anomalies in the hands, feet, and genitals, as well as a condition known as complex split hand defect. The severity of these features can vary among affected individuals.
6q25-qter deletion syndrome
Key features of this syndrome include difficulties feeding, hydrocephalus, seizures, heart conditions, developmental delay, and unusual facial features. Additionally, individuals with this syndrome may have a vision impairment, minor genital anomalies, spinal defect, cleft palate and swelling of one or both kidneys. The severity of these features can vary among affected individuals.
6q26-q27 deletion syndrome
Key features of this syndrome include feeding difficulties, intellectual disability, distinct facial features, seizures, weak muscle tone (hypotonia), minor anomalies of the hands and feet, multiple malformations, and brain abnormalities.
7q deletion
7q deletion is also known as Familial monosomy 7 syndrome. The severity of the condition and the signs and symptoms depend on the size and location of the deletion and which genes are involved. Key features of this deletion include developmental delay, intellectual disability, behavioral problems, and distinctive facial features. The deletion of several genes of chromosome 7 may cause the development of myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML). Symptoms typically include small red or purple spots on the skin (petechiae), easy bruising, or anemia. Rapid progression is common, and prognosis is generally poor. Most cases are not inherited, but people can pass the deletion on to their children.
7q11.23 deletion syndrome
Key features of this syndrome include with increased risk for epilepsy, neurodevelopmental disorders (including developmental delay and intellectual disabilities of variable severity), learning difficulties, and neurobehavioral abnormalities (autism spectrum disorder, hyperactivity, impulsivity, aggression, self-abusive behaviors, and depression).
7q21-q32 deletion syndrome
Key features of this syndrome include mild to severe developmental delay and difficulties with learning. While most babies are born with a healthy heart, defects have been found in more than one-third. And while most children do not develop epilepsy, almost one in three babies or children has had seizures. Other symptoms can include specific social behavior, which can include autism spectrum disorder and difficulties with language, speech, sleeping and feeding.
7q31-q32 deletion syndrome
Deletions in region 7q31-q32 are associated with a severe communication disorder with evidence of oromotor dyspraxia (difficulties in coordinating muscle movements needed to pronounce words), dysmorphic features, and mild developmental delay.
8p23.1 deletion syndrome
The clinical manifestations are variable and do not depend on the size of the deletion since it is the same in most patients. Key features of this syndrome include low birth weight, postnatal growth deficiency, mild intellectual deficit, psychomotor retardation, poor speech, seizures, behavioral problems such as hyperactivity and impulsiveness, craniofacial abnormalities (microcephaly, high and narrow forehead, broad nasal bridge, epicanthic folds, high arched palate, short neck, and low set unusually shaped ears), and congenital heart defects.
8p23.1 duplication syndrome
Key features of this syndrome include mild to moderate developmental delay, intellectual disability, mild facial dysmorphism (incl. prominent forehead, arched eyebrows, broad nasal bridge, upturned nares, cleft lip and/or palate) and congenital cardiac anomalies (e.g., atrioventricular septal defect). Other reported features include macrocephaly, behavioral abnormalities (e.g., attention deficit disorder), seizures, hypotonia and ocular and digital anomalies (poly/syndactyly). The prevalence of 8p23.1 duplication syndrome is 1 to 9 in 100,000.
Langer-Giedion syndrome (LGS)
Langer-Giedion syndrome, also known as Trichorhinophalangeal syndrome type 2, is a rare multisystem disorder caused by a deletion on chromosome 8, which often includes the TRPS1, RAD21 and EXT1 gene. Key features of this syndrome include intellectual deficit and numerous other abnormalities including excess folds of skin, multiple bony growths (exostoses), short stature, sparse and depigmented scalp hair, typical facial characteristics (broad eyebrows, especially the medial portion, broad nasal ridge and tip, underdeveloped nasal alae, long philtrum, thin upper lip vermilion, and protruding ears), limb anomalies, and cone-shaped phalangeal epiphyses (the growing ends of the bones in the fingers). The range and severity of symptoms vary greatly from person to person.
8q22.1 deletion syndrome
8q22.1 deletion syndrome, also known as Nablus mask-like facial syndrome, is characterized by a mask-like facial appearance. Facial features include narrowing of the eye opening (blepharophimosis), tight appearing glistening facial skin, and flat and broad nose. Other features include malformed ears, unusual scalp hair pattern, permanently bent fingers and toes (camptodactyly), joint deformities (contractures) that restrict movement in the hands and feet, unusual dentition, mild developmental delay, undescended testicles in males (cryptorchidism), and a happy disposition.
8q22.1 duplication syndrome
Also known as Leri pleonosteosis chromosome duplication syndrome, this condition includes broadening and deformity of the thumbs and great toes in a valgus position (a 'spade-shaped' appearance), flexion contracture of the interphalangeal joints, generalized limitation of joint mobility, short stature, and often mongoloid facies. Additional malformations include genu recurvatum, enlargement of the posterior neural arches of the cervical vertebrae and thickening of the palmar and forearm fasciae.
8p duplication syndrome
Key features of this syndrome include a highly variable phenotype ranging from no dysmorphic features and only mild intellectual disability to patients with severe developmental delay, weak muscle tone or "floppiness" in newborn infants, short stature, profound intellectual disability, mild dysmorphic features, and structural brain abnormalities. Autism, epilepsy, and spastic paraplegia (a neurological disorder characterized by weakness and stiffness in the muscles below the waist) have also been reported.
8q duplication syndrome
The symptoms and severity depend on the size and location of the duplication, which genes are involved, and whether other chromosome abnormalities are also present. Key features of this syndrome include developmental delay, learning difficulties, congenital heart defects, skeletal abnormalities, genital or urinary abnormalities, and distinctive facial features.
9p deletion syndrome
The severity and the signs and symptoms depend on the size and location of the deletion and which genes are involved. Key features of this syndrome include developmental delay, low muscle tone (hypotonia), distinctive facial features (trigonocephaly, midface hypoplasia, up-slanting palpebral fissures, dysplastic small ears, flat nasal bridge with anteverted nostrils and long philtrum, micrognathia, choanal atresia, short neck), heart conditions, scoliosis, and/or genital abnormalities.
9p duplication
Key features of this duplication include intellectual disability, craniofacial dysmorphism (e.g. abnormally small head size (microcephaly), large anterior fontanel, widely spaced eyes (hypertelorism), strabismus, down slanting eyes, malformed, low-set, protruding ears, bulbous nose, large mouth, down-turned corners of mouth, abnormally small jaw), digital anomalies (brachydactyly and clinodactyly), and short stature. Less frequently patients present with cardiopathy and renal, skeletal, and central nervous system malformations.
DiGeorge syndrome 2
DiGeorge syndrome 2 involves cardiac malformations, hypoparathyroidism, T-cell immunodeficiency, and facial dysmorphism. These features overlap with the anomalies reported in the 22q11.2 deletion syndrome (DiGeorge syndrome). In addition, other common features also include an abnormally shaped skull, abnormally small head size (microcephaly), hand and foot abnormalities, a long face, a high forehead, a broad nasal bridge, genitourinary anomalies, severe psychomotor retardation, and hearing loss, resulting in a clinical picture that clearly differs from that of the DiGeorge syndrome. However, because of several similarities in the abnormalities observed in individuals with deletions 22q11.2, the syndrome is often referred to as DiGeorge syndrome 2.
10q22.3-q23.2 deletion syndrome
Key features of this syndrome include dysmorphic facial features, developmental delay, and multiple congenital anomalies. More severe cases that include the deletion of the PTEN gene have a more severe phenotype with infantile/juvenile polyposis (development of multiple polyps in the gastrointestinal tract), abnormally large head (macrocephaly), dysmorphic facial features, and developmental delay.
10q26 deletion syndrome
The severity of the condition and the signs and symptoms depend on the size and location of the deletion and which genes are involved. Key features of this syndrome include facial dysmorphism, mild to moderate intellectual disability, growth problems, and developmental delay. Affected individuals may experience seizures, attention-deficit/hyperactivity disorder (ADHD), impulsivity, or exhibit autistic behaviors. Facial features may include a prominent or beaked nose, a broad nasal bridge, a small lower jaw, malformed ears that are low set, a thin upper lip, and an abnormally small head. Many affected individuals have widely spaced eyes (hypertelorism) and squint. Less common signs include skeletal and heart abnormalities, breathing problems, recurrent infections, kidney and genital abnormalities, or hearing and vision problems. Most cases are not inherited, but people can pass the deletion on to their children.
10p12-p11 deletion syndrome
Key features include developmental delay apparent in infancy or early childhood and associated with characteristic dysmorphic facial features, such as broad forehead, depressed nasal bridge with bulbous nasal tip, and deep-set eyes. Other common features include hypotonia, speech delay, mild to moderate intellectual disability, abnormal behavior (autistic, aggressive, hyperactive), and congenital heart and brain anomalies. Most patients also have gastrointestinal and mild eye abnormalities.
10p duplication
10p duplication means that a part of the short arm of chromosome 10 is duplicated. The severity and the symptoms of chromosome 10p duplication depend on the size and location of the duplication and which genes are involved. Key features of this duplication include delay of development, motor skills, or growth, short stature, low muscle tone, abnormalities of the feet (such as clubfeet), cleft lip and/or cleft palate, and distinctive facial features. Other signs and symptoms may include seizures, a heart defect, or other birth defects.
11p13 deletion syndrome (WAGR syndrome)
WAGR is an acronym for Wilms tumor, Aniridia, Genitourinary problems, and Range of developmental delays. Key features of this syndrome include complete or partial congenital aniridia (underdevelopment or absence of the iris, which can reduce the sharpness of vision and increase sensitivity to light) and associated eye abnormalities, genitourinary anomalies (such as undescended testicles or hypospadias in males, or internal genital or urinary anomalies in females), variable degrees of intellectual disability and an increased risk of developing Wilms tumors. A minority of patients develop kidney failure, and many patients develop obesity. However, WAGR syndrome with childhood-onset obesity is called WAGRO syndrome.
11p11.2 deletion syndrome (Potocki-Shaffer syndrome)
Key features of this syndrome include openings in the two bones that form the top and sides of the skull (enlarged parietal foramina), multiple benign (non-cancerous) bone tumors called exostoses, intellectual disability, developmental delay, a distinctive facial appearance, autism spectrum disorder, and problems with vision and hearing. In some cases, individuals with the syndrome may have a defect in the heart, kidneys, or urinary tract.
Jacobsen syndrome
Jacobsen syndrome is a rare genetic syndrome caused by a deletion of several genes on the terminal end of the long arm of chromosome 11. Signs and symptoms vary among affected people. Key features of this syndrome often include Paris-Trousseau syndrome (a bleeding disorder), distinctive facial features, delayed development of motor skills and speech, and cognitive impairment. Other features may include compulsive behavior, attention deficit-hyperactivity disorder (ADHD), autism, seizures, congenital heart defects, short stature, and/or skeletal abnormalities.
11q23 deletion syndrome
Key features of this syndrome, also known as Paris-Trousseau thrombocytopenia, include mild bleeding tendency, variable thrombocytopenia (fluctuating low platelet counts), distinctive facial features, abnormal giant alpha-granules in platelets and dysmegakaryopoiesis. Other frequent symptoms include intellectual disability and abnormalities of the cardiovascular system.
12q14 microdeletion syndrome
Key features of this syndrome include a mild form of mental disability, early developmental disorders, and osteopoikilosis (excessive bone density leading to pain). Patients are characterized by short stature.
12p12.1 microdeletion syndrome
Key features of this syndrome, also known as Lamb-Shaffer syndrome, include challenges with learning, delays in overall development with language impairment, unusual behaviors, and mild facial dysmorphism, like a broad forehead, eyes that slant downward, and low-set ears. Other possible features include skeletal abnormalities, crossed eyes, optic nerve hypoplasia, and problems with the brain's structure.
12p duplication
The severity of the condition and the signs and symptoms depend on the size and location of the duplication and which genes are involved. Key features of this condition include macrocephaly (unusually large head), low muscle tone, characteristic facial features (including frontal bossing, round face, full cheeks, low-set ears, broad nasal bridge, short nose with anteverted nares, long philtrum, thin upper lip vermilion, and everted, thick lower lip), developmental delay and intellectual disability. Most cases are not inherited, but people can pass the duplication on to their children.
13q14 deletion syndrome
Key features of this syndrome include retinoblastoma, developmental delay, variable degrees of intellectual disability, delayed language development, characteristic facial features, including a high forehead, a noticeable space between the nose and upper lip, large mouth with thin upper lip and thick, everted lower lip, and ears that slant forward. Other features reported include high birth weight, high birth weight, large head size (macrocephaly), tumor of the pineal gland (pinealoma), enlarged liver (hepatomegaly), hernia in the groin area and undescended testicles.
Distal chromosome 13q deletion syndrome
Key features of this syndrome include varying degrees of intellectual disability and developmental delay, as well as central nervous system malformations, ocular abnormalities and craniofacial dysmorphism (abnormally small and triangular-shaped head, large, and malformed ears, broad prominent nasal bridge, and small/underdeveloped lower jaw). Cardiac, genitourinary, gastrointestinal, and skeletal manifestations have also been reported.
14q11-q22 deletion syndrome
Key features of this syndrome include an abnormally small head, low muscle tone, poor growth, intellectual disability with poor eye contact, hypoplasia of the corpus callosum and dysmorphic features, including a short nose, long space between the nose and upper lip, and flat nasal bridge.
14q22 deletion syndrome (Frias syndrome)
Key features of this syndrome include ocular anomalies (anophthalmia/microphthalmia, ptosis, hypertelorism, exophthalmos), pituitary anomalies (pituitary hypoplasia/aplasia with growth hormone deficiency and growth retardation) and hand/foot anomalies (polydactyly, short digits, pes cavus). Other clinical features may include muscular hypotonia, psychomotor development delay/intellectual disability, dysmorphic signs (facial asymmetry, microretrognathia, high-arched palate, ear anomalies), congenital genitourinary malformations, and hearing impairment.
Proximal chromosome 14q deletion syndrome
Key features of this syndrome include developmental delay, reluctance, or inability to feed, low muscle tone, abnormally small head, small lower jaw, minor anomalies of genitals (more common in boys), low birth weight, respiratory distress, and partial or complete split in the roof of the mouth (cleft palate).
14q duplication syndrome
Key features of this syndrome include growth retardation and low birth weight, low muscle tone, developmental delay, intellectual disability, short stature, abnormally small head, facial dysmorphism (frontal bossing, widely spaced eyes (hypertelorism), bulbous nose, small/underdeveloped jaw, sparse hair and eyebrows), congenital heart defects, muscle stiffness, and hyperreflexia.
Angelman syndrome/Prader-Willi syndrome (15q11-q13 deletion syndrome)
Prader-Willi syndrome (PWS) is a genetic syndrome that causes difficulty feeding and failure to thrive in infancy, with obesity, developmental delay, and other medical problems as the child gets older. It affects approximately 1 in 10,000 to 1 in 25,000 newborns. NIPT is able to detect PWS caused by a deletion, which accounts for approximately 70% of cases; the remaining cases are caused by different underlying molecular mechanisms. Deletion in PWS is on the paternally inherited chromosome.
Angelman syndrome (AS) is a rare genetic syndrome that includes intellectual disability and other serious medical problems. It affects approximately 1 in 12,000 to 1 in 20,000 newborns. NIPT is able to detect AS caused by a deletion, which accounts for approximately 68% of cases; the remaining cases are caused by different underlying molecular mechanisms. Deletion in AS is on the maternally inherited chromosome.
People with Prader-Willi syndrome have severe hypotonia and feeding difficulties in early infancy, followed in later infancy or early childhood by excessive eating and gradual development of morbid obesity. Motor milestones and language development are delayed. All individuals have some degree of cognitive impairment and may also have behavior problems. Prenatal ultrasounds are usually normal; however, decreased fetal movement and breech position may be seen. A normal lifespan is expected.
Common features in individuals with Angelman syndrome include intellectual disability, severe developmental delay, speech impairment, ataxia, seizures, and dysmorphic features. Prenatal ultrasound is usually normal. A normal lifespan is expected.
15q26-qter deletion syndrome
Key features of this syndrome include variable degrees of intellectual disability, delayed psychomotor development, pre-and postnatal growth restriction, and some mild changes in the face, like a smaller head and a triangular-shaped face. Other possible features include distinct facial characteristics, hand and foot anomalies, heart problems, and autism spectrum disorder.
Levy-Shanske syndrome
Levy-Shanske syndrome is a rare genetic syndrome caused by a tetrasomy of chromosome 15q26-qter. Key features of this syndrome include pre- and post-natal overgrowth, renal anomalies, facial dysmorphism (long thin face, prominent forehead and chin, down-slanting eyelids, prominent nose with broad nasal bridge), mild to severe learning difficulties and behavioral problems. Additional clinical signs may include an abnormally large head and craniosynostosis.
15q14 deletion syndrome
Key features of this syndrome include delayed psychomotor development, short stature, facial dysmorphism, and variable degrees of intellectual disability. Dysmorphic features include bitemporal narrowing, high forehead, short space between the nose and upper lip, pointed chin and dysmorphic ears. A split in the roof of the mouth (cleft palate) has been described in all reported cases, while congenital heart defects or epilepsy have been observed in patients with large deletions.
15q24 microdeletion syndrome
Key features of this syndrome include pre- and post-natal growth retardation, intellectual disability, distinct facial features, and genital, skeletal, and digital anomalies.
15q26 overgrowth syndrome
Key features of this syndrome include renal anomalies (45%), such as horseshoe kidney and renal agenesis (one or both fetal kidneys fail to develop). Inadequate functioning of the kidneys, backward flow of urine, polycystic kidney and right renal pelvic duplication are common as well. People with this syndrome can also have intellectual disabilities, experience developmental delays, and have a tall stature. Additional features may include craniosynostosis and macrocephaly.
Distal chromosome 15q deletion
Key features of this deletion include pre- and postnatal growth restriction, developmental delay, variable degrees of intellectual disability, hand and foot anomalies and mild craniofacial dysmorphism. Other possible features include swelling in newborns, heart problems, skin issues, a widened aorta, and being on the autism spectrum.
16p12.2-p11.2 deletion syndrome
Key features of this syndrome include developmental delay, distinct facial features (flat facies, down slanting eyelids, low-set, and malformed ears), feeding difficulties, recurrent ear infections, and cognitive impairment. Additional features, such as heart defects, orofacial clefting, and short stature can be observed.
16p12.2-p11.2 duplication syndrome
Key features of this syndrome include developmental/psychomotor delay (particularly of speech), intellectual disability, autism spectrum disorder and/or obsessive and repetitive behavior, behavioral problems (such as aggression and outbursts), and dysmorphic facial features. Other common features include finger or hand anomalies, short stature, having a smaller head, and a slim build.
16p13.3 deletion syndrome
This deletion results in a severe form of Rubinstein-Taybi syndrome. Some researchers believe that it is a unique emerging syndrome. Key features of this syndrome include failure to thrive, hypotonia (reduced muscle tone), short stature, microcephaly (unusually small head), characteristic facial features, mild to moderate intellectual disability, organ anomalies (i.e. heart and/or kidney problems), and vulnerability to infections.
16p13.3 duplication syndrome
Key features of this syndrome include mild to moderate intellectual deficit and developmental delay (particularly speech), normal growth, short, proximally implanted thumbs and other hand and feet malformations, mild joint stiffness, characteristic facial features, attention deficit, autism spectrum disorders, and underlying health problems such as heart conditions. Other reported issues include undescended testicles, a hernia in the groin area, and behavioral problems.
Proximal chromosome 16q duplication
Key features of this duplication include developmental delay, learning difficulties, speech delay, minor anomalies of the hands and/or feet, abnormal teeth development (often resulting in small teeth), and attention/behavior difficulties. Despite these challenges, affected individuals are usually healthy and do not have major birth defects.
Smith-Magenis syndrome
Smith-Magenis syndrome (SMS) is a rare genetic syndrome caused by a deletion on the short arm of chromosome 17. Although this region contains multiple genes, the loss of one particular gene, RAI1, is responsible for most of the features of the condition. In about 10% of cases, SMS is caused by a genetic change in the RAI1 gene. Key features of this syndrome include mild to moderate intellectual disability, delayed speech and language skills, distinctive facial features, sleep disturbances, and behavioral problems. The prevalence of Smith-Magenis syndrome is 1 in 15,000-25,000.
17p13.3 deletion syndrome (Miller-Dieker syndrome)
Key features of this syndrome include lissencephaly (a rare brain development disorder characterized by smooth or absent brain folds and abnormally thickened cerebral cortex) and an abnormally small head which causes severe intellectual disability, developmental delay, seizures, abnormal muscle stiffness, weak muscle tone, and feeding difficulties. Seizures usually begin before six months of age, and some occur from birth. Patients also present with distinctive facial features. Some individuals may also be smaller in size, have heart problems, or face serious breathing issues. Life expectancy is greatly reduced, and death most often occurs in early childhood.
Potocki-Lupski syndrome
Potocki-Lupski syndrome is a rare genetic syndrome caused by duplication on the short arm of chromosome 17. Key features of this syndrome include some degree of developmental delay (primarily cognitive and language deficits), low muscle tone, poor feeding, mild-moderate intellectual delay, and failure to thrive during infancy. In addition, many individuals display some behaviors commonly associated with autism spectrum disorders. Structural cardiovascular anomalies and sleep disturbance have also been frequently reported.
17p13.3 duplication syndrome
Key features of this syndrome include intellectual disability, autism, and structural brain abnormalities. Duplications might involve the LIS1 and/or the YWHAE gene. When both genes are involved, the clinical signs include distinct facial features and frequent structural brain abnormalities. Individuals with LIS1 duplications have subtle brain defects, as well as neurobehavioral disorders, including delayed development, mental retardation, and attention deficit hyperactivity disorder. YWHAE duplications cause features like larger size at birth, unusual facial appearance, and mild delays in development.
Yuan-Harel-Lupski syndrome
Yuan-Harel-Lupski syndrome is a rare genetic syndrome caused by a duplication on the short arm of chromosome 17. Key features of this syndrome include delays in overall development and problems with nerves in the limbs (peripheral neuropathy) that start early in life. It includes characteristics of two other conditions, Charcot-Marie-Tooth disease type 1A and Potocki-Lupski syndrome. The combined effects of these conditions can lead to more serious symptoms, such as an early start of nerve problems and involvement of both the central and peripheral nervous systems. Other features include weak muscle tone, feeding difficulties, and failure to thrive, as well as gait impairment (a walking disorder), sensory loss, reduced or absent deep tendon reflexes of the ankles, and foot deformities. Facial dysmorphism, cardiac and renal anomalies, and syringomyelia (a neurological disorder in which a fluid-filled cyst (syrinx) forms within the spinal cord) have also been reported.
17p duplication
The severity of the condition and the signs and symptoms depend on the size and location of the duplication and which genes are involved. Key features of this duplication include pre- and post-natal growth retardation, feeding difficulties, developmental delay and learning difficulties, behavior difficulties such as hyperactivity and autistic features, speech and language delay, weak muscle tone, digital abnormalities, congenital heart defects, scoliosis, sleep apnea, and distinctive facial features.
18p deletion syndrome
Key features of this syndrome include mental disabilities, growth disturbances, head and face anomalies (round face, enlarged oral cavity), abnormal ears and anomalies of the limbs, genitalia, brains, eyes, heart, and teeth.
Distal chromosome 18q deletion syndrome
Key features of this syndrome are highly variable and include mental disorders, short stature, weak muscle tone, hearing disorders, feet deformities, pointed toes, and mild facial differences such as deep-set eyes, a flat or sunken appearance of the middle of the face (midface hypoplasia), a wide mouth, and prominent ears. Eye movement disorders and other vision problems, cleft palate, an underactive thyroid gland (hypothyroidism), congenital heart defects, kidney problems, genital abnormalities, and skin problems may also occur in this syndrome. Distal chromosome 18q deletion syndrome can also affect the nervous system and cause neurological problems.
Alagille syndrome 1
Alagille syndrome 1 (AGS type 1) is a rare genetic syndrome caused by a deletion of p12 region of chromosome 20. Around 7% of all Alagille syndromes (AGS type 1) are caused by this deletion that includes JAG1 gene. In more than 90 percent of cases, mutations in the JAG1 gene cause Alagille syndrome (AGS type 1). AGS type 2 is due to NOTCH2 gene mutations (on chromosome 1p12). Key features of this syndrome include liver damage caused by abnormalities in the bile ducts, and several heart problems, but it can also affect other parts of the body. People with Alagille syndrome may have distinctive facial features including a broad, prominent forehead, deep-set eyes, and a small, pointed chin. The disorder may also affect the blood vessels within the brain, spinal cord, and kidneys. Affected individuals may have an unusual butterfly shape of the bones of the spinal column (vertebrae). The prevalence of Alagille syndrome is approximately 1 in 70,000.
20p duplication
The severity of the condition and the signs and symptoms depend on the size and location of the duplication and which genes are involved. In general, smaller duplications are less severe than larger duplications. Key features of this duplication include intellectual disability, developmental delay, speech delay, poor coordination, dental problems, spinal bone abnormalities, distinctive facial features, and heart problems.
21q22 deletion
Key features of this deletion include intrauterine and postnatal growth retardation, an abnormally small head, an enlarged or protruding back part of the skull, and facial dysmorphism characterized by up or down-slanted small eyelids, prominent nasal bridge with a broad nose, and large ears. Multiple malformations including structural brain malformations and heart defects are also frequently observed. Other common features include severe intellectual deficit, stiff joints, abnormal muscle tone, respiratory infections, and seizures. Specific blood disorders (e.g. thrombocytopenia, myelodysplasia) have been reported.
22q11.2 deletion syndrome (DiGeorge syndrome)
DiGeorge syndrome occurs in approximately 1 in 4,000 live births. The clinical phenotype is extremely variable, ranging from mild to severe. Key features include learning and behavioral problems, mild intellectual disability, palatal anomalies, and dysmorphic features. Cardiac defects are seen in 85% of cases. Renal and brain anomalies may also be present. Some patients have immune deficiency due to thymic hypoplasia, thus increasing the risk for infection, as well as hypoparathyroidism, which causes hypocalcemia. Life span is usually normal but can vary depending on the severity of features. Prenatal ultrasound may help to identify structural anomalies; however, a normal ultrasound does not exclude the condition.
As the signs and symptoms of 22q11.2 deletion syndrome are very diverse, different groupings of features were previously identified as separate conditions, such as DiGeorge syndrome, velocardiofacial (Shprintzen) syndrome and conotruncal anomaly face syndrome, and in some cases Opitz G/BBB syndrome and Cayler cardiofacial syndrome. However, the discovery of their common genetic basis led doctors to group them together into one single syndrome with many possible signs and symptoms, called 22q11.2 deletion syndrome.
Xp11.23-p11.22 duplication syndrome
Key features of this syndrome include borderline to severe mental retardation, speech delay, EEG abnormalities, and early puberty. Additionally, individuals may be significantly overweight and display lower-extremity anomalies, fifth-toe hypoplasia, and syndactyly. Some affected individuals also show an absence of the corpus callosum, which is the band of nerve fibers connecting the two hemispheres of the brain.
Xp21 deletion syndrome
Xp21 deletion syndrome is a rare genetic syndrome caused by a deletion of p21 region of chromosome X that usually affects males. Key features of this syndrome include complex glycerol kinase deficiency that can lead to congenital adrenal hypoplasia (ACH), intellectual disability, and/or Duchenne muscular dystrophy (DMD). People with this syndrome may have high levels of glycerol in their blood and urine, and the symptoms can vary depending on which genes are missing and how many are involved.
Xq27.3-q28 duplication syndrome
Key features of this syndrome include mild intellectual disability, facial dysmorphism (deep-set eyes, bulbous nasal tip, and thin lips), short stature, and primary gonadal failure which manifests as high-pitched voice, sparse body hair, abdominal obesity, and hypogonadism in affected males. Female carriers may have short stature and experience early menopause.
Xq21 deletion syndrome
Xq21 deletion syndrome, also known as X-linked retinal dystrophy, is a rare genetic syndrome caused by a deletion of q21 region of chromosome X. Key features of this syndrome include choroideremia, which causes progressive nyctalopia (night blindness) and eventual central blindness in males. Additionally, patients may experience moderate intellectual disability, congenital mixed deafness (involving both sensorineural and conductive components), and obesity. Female carriers show typical retinal changes indicative of the choroideremia carrier state.
Xq22.3 deletion syndrome
Xq22.3 deletion syndrome, also known as X-linked Alport syndrome-diffuse leiomyomatosis (XLAS-DL), is a rare genetic syndrome caused by a deletion of the q22.3 region of chromosome X. Key features of this syndrome include glomerular disease symptoms, such as blood in urine, increasing protein in urine, and kidney problems, which typically appear later in childhood. In males, the disease tends to be more severe and progresses more rapidly. Patients also experience hearing loss and ocular anomalies, such as lens problems, cataracts, cloudy corneas, and retina problems. Diffuse leiomyomatosis (a rare condition characterized by the abnormal growth of smooth muscle tissue) is severe in both males and females. It mainly affects the oesophagus, causing trouble swallowing, vomiting after meals, and chest or stomach pain. It also affects the airways, leading to breathing problems, coughing, noisy breathing, and frequent lung infections from breathing in food. Affected females may experience an enlarged clitoris, as well as varying degrees of impact on their outer genitalia and uterus.
60 syndromes of deletion and duplication (NIPT Plus)
Alpha Thalassemia, mental retardation syndrome
Causes severe mental and physical disability with facial anomalies. The characteristic facial features include wide-set eyes, epicanthic fold (upper eyelid covering the inner corner of the eye), a small triangle-shaped nose, flat face, and microcephaly. The majority of patients also have genital anomalies and a severe form of anaemia.
Androgen insensitivity syndrome (AIS)
Androgen insensitivity syndrome is a condition that affects sexual development before birth and during puberty. People with this condition are genetically male, with one X chromosome and one Y chromosome in each cell. Because their bodies are unable to respond to certain male sex hormones (called androgens), they may have mostly female external sex characteristics or signs of both male and female sexual development.
Angelman syndrome/Prader-Willi syndrome (15q11-q13 deletion syndrome)
Prader-Willi syndrome (PWS) is a genetic syndrome that causes difficulty feeding and failure to thrive in infancy, with obesity, developmental delay, and other medical problems as the child gets older. It affects approximately 1 in 10,000 to 1 in 25,000 newborns. NIPT is able to detect PWS caused by a deletion, which accounts for approximately 70% of cases; the remaining cases are caused by different underlying molecular mechanisms. Deletion in PWS is on the paternally inherited chromosome.
Angelman syndrome (AS) is a rare genetic syndrome that includes intellectual disability and other serious medical problems. It affects approximately 1 in 12,000 to 1 in 20,000 newborns. NIPT is able to detect AS caused by a deletion, which accounts for approximately 68% of cases; the remaining cases are caused by different underlying molecular mechanisms. Deletion in AS is on the maternally inherited chromosome.
People with Prader-Willi syndrome have severe hypotonia and feeding difficulties in early infancy, followed in later infancy or early childhood by excessive eating and gradual development of morbid obesity. Motor milestones and language development are delayed. All individuals have some degree of cognitive impairment and may also have behavior problems. Prenatal ultrasounds are usually normal; however, decreased fetal movement and breech position may be seen. A normal lifespan is expected.
Common features in individuals with Angelman syndrome include intellectual disability, severe developmental delay, speech impairment, ataxia, seizures, and dysmorphic features. Prenatal ultrasound is usually normal. A normal lifespan is expected.
Bannayan-Riley-Ruvalcaba syndrome (BRRS)
Bannayan-Riley-Ruvalcaba syndrome is a genetic condition characterised by a large head (macrocephaly), multiple noncancerous tumours and tumour-like growths called hamartomas, and dark freckles on the penis in males. The signs and symptoms of this syndrome are present from birth or become apparent in early childhood.
Branchiootorenal dysplasia syndrome (BOR)/Melnick-Fraser syndrome
Branchiootorenal (BOR) syndrome is a condition that disrupts the development of tissues in the neck and causes the malformation of ears and kidneys.
Cat eye syndrome (CES)
Cat eye syndrome (CES) is a rare chromosomal disorder with a highly variable clinical presentation. Most patients have multiple malformations affecting the eyes (iris coloboma), ears (preauricular pits and/or tags), anal region (anal atresia), heart, and kidneys. Intellectual disability is usually mild or borderline normal.
Chromosome 10q deletion syndrome
This deletion is accompanied by facial asymmetry, pronounced nose, narrow upper lip, low birth weight, short stature, fifth finger clinodactyly and male cryptorchidism (absence of one or both testes). People with deletion 10q have different forms of learning difficulties and delayed development.
Chromosome 10q22.3–q23.31 microdeletion syndrome
Recurrent deletions of chromosome 10q22.3–q23.31 have been associated with dysmorphic facial features, developmental delay, and multiple congenital anomalies.
Chromosome 18p deletion syndrome
The main clinical features of this deletion are mental disabilities, growth disturbances, head and face anomalies (round face, enlarged oral cavity), abnormal ears and anomalies of the limbs, sexual organs, brains, eyes, heart, and teeth.
Distal chromosome 18q deletion syndrome
Key features of this syndrome are highly variable and include mental disorders, short stature, weak muscle tone, hearing disorders, feet deformities, pointed toes, and mild facial differences such as deep-set eyes, a flat or sunken appearance of the middle of the face (midface hypoplasia), a wide mouth, and prominent ears. Eye movement disorders and other vision problems, cleft palate, an underactive thyroid gland (hypothyroidism), congenital heart defects, kidney problems, genital abnormalities, and skin problems may also occur in this syndrome. Distal chromosome 18q deletion syndrome can also affect the nervous system and cause neurological problems.
Cornelia de Lange syndrome (CDLS)
Cornelia de Lange syndrome is characterised by facial dysmorphism, elevated eyebrows, a unibrow (synophrys), curled nose, narrow lips, enlarged middle part of the upper lip, reduced lower jaw growth. Clinical features include developmental delays, growth and cognitive impairment, and limb malformations.
Cowden syndrome (CD)
Cowden syndrome characteristics include benign lesions in the common tissue (skin, mucous membrane, chest and thyroid), on the face (near and in the mouth) and on the limbs.
Cri-du-chat syndrome (5p deletion syndrome)
5p deletion syndrome, also known as Cri-du-chat syndrome, is a genetic syndrome characterized by birth defects, intellectual disability, and other serious medical issues. Key features of this syndrome include significant intellectual disability, speech delay, cat-like cry, dysmorphic features, cardiac defects, and microcephaly. There is a 10% mortality rate in the first year. Approximately 1 in 20,000 to 1 in 50,000 live births have this condition.
Dandy-Walker syndrome (DWS)
Dandy-Walker syndrome is a rare genetic syndrome caused by a deletion in chromosome region 3q22-q24. Key features of this syndrome include affected brain development, specifically the part called the cerebellum, which is responsible for coordinating movement. The syndrome’s main feature is the enlarged fourth ventricle in the brain. Up to half of the affected individuals have intellectual disability, delay in the development of motor skills, muscle stiffness, and seizures. The incidence is approximately 1 in 25,000-35,000.
DiGeorge syndrome 2
DiGeorge syndrome 2 involves cardiac malformations, hypoparathyroidism, T-cell immunodeficiency, and facial dysmorphism. These features overlap with the anomalies reported in the 22q11.2 deletion syndrome (DiGeorge syndrome). In addition, other common features also include an abnormally shaped skull, abnormally small head size (microcephaly), hand and foot abnormalities, a long face, a high forehead, a broad nasal bridge, genitourinary anomalies, severe psychomotor retardation, and hearing loss, resulting in a clinical picture that clearly differs from that of the DiGeorge syndrome. However, because of several similarities in the abnormalities observed in individuals with deletions 22q11.2, the syndrome is often referred to as DiGeorge syndrome 2.
Distal arthrogryposis type 2B (DA2B)
Common clinical features of type 2B distal arthrogryposis include clenched fist, overlapping fingers, camptodactyly, ulnar deviation, and positional foot deformities from birth. Patients with this syndrome are recognised by nasolabial folds, down slanting palpebral fissures, and small mouth.
Duchenne and Becker muscular dystrophy (DMD/BMD)
Muscular dystrophies are a group of genetic conditions characterised by progressive muscle weakness and wasting (atrophy). The Duchenne and Becker types of muscular dystrophy are two related conditions that primarily affect skeletal muscles, which are used for movement, and heart (cardiac) muscle. These forms of muscular dystrophy occur almost exclusively in males.
Dyggve-Melchior-Clausen syndrome (DMC)
DMC syndrome is a rare, progressive genetic condition characterised by abnormal skeletal development, microcephaly, and intellectual disability. Only about 100 cases have been reported to date.
Feingold syndrome
Feingold syndrome is an autosomal dominant disorder characterised by variable combinations of microcephaly, limb malformations, oesophageal and duodenal atresia, and learning disability/intellectual disability.
Holoprosencephaly type 1 (HPE1)
Holoprosencephaly (HPE1) is the most common structural malformation of the human forebrain and occurs after failed or abbreviated midline cleavage of the developing brain during the third and fourth week of gestation.
Holoprosencephaly type 4 (HPE4)
Holoprosencephaly type 4 is characterised by severe facial and brain anomalies. The degree of disability depends on the degree of brain injury. Severe forms of the syndrome are often fatal.
Holoprosencephaly type 6 (HPE6)
Type 6 holoprosencephaly is characterised by deformation of the brain and face.
Jacobsen syndrome
Jacobsen syndrome is a rare genetic syndrome caused by a deletion of several genes on the terminal end of the long arm of chromosome 11. Signs and symptoms vary among affected people. Key features of this syndrome often include Paris-Trousseau syndrome (a bleeding disorder), distinctive facial features, delayed development of motor skills and speech, and cognitive impairment. Other features may include compulsive behavior, attention deficit-hyperactivity disorder (ADHD), autism, seizures, congenital heart defects, short stature, and/or skeletal abnormalities.
Langer-Giedion syndrome (LGS)
Langer-Giedion syndrome, also known as Trichorhinophalangeal syndrome type 2, is a rare multisystem disorder caused by a deletion on chromosome 8, which often includes the TRPS1, RAD21 and EXT1 gene. Key features of this syndrome include intellectual deficit and numerous other abnormalities including excess folds of skin, multiple bony growths (exostoses), short stature, sparse and depigmented scalp hair, typical facial characteristics (broad eyebrows, especially the medial portion, broad nasal ridge and tip, underdeveloped nasal alae, long philtrum, thin upper lip vermilion, and protruding ears), limb anomalies, and cone-shaped phalangeal epiphyses (the growing ends of the bones in the fingers). The range and severity of symptoms vary greatly from person to person.
Leukodystrophy with 11q14.2–q14.3
Microdeletion 11q14.2–q14.3 can cause leukodystrophy, which is accompanied by epileptic seizures in infants, severe psychomotor disabilities, growth and developmental disorders, microcephaly, microphallus, structural brain disorders and facial anomalies (e.g., round nasal tip, forehead and eyelid deformities). Patients with leukodystrophy die during childhood.
Mental retardation, X-linked growth hormone deficiency (MRGH)
Mental retardation, X-linked, with isolated growth hormone deficiency: a disorder characterised by the association of variable degrees of intellectual disability with panhypopituitarism, variable combinations of hypothyroidism, delayed pubertal development, and short stature due to growth hormone deficiency.
Microphthalmia, syndromic 6, pituitary hypoplasia
Patients with this syndrome are characterised by the absence of one or both eyes, a brachycephaly (shorter skull), retrognathia (lower jaw deformity), small ears, anomalies of the fingers, an underdevelopment of the external genitalia, and a hypoplastic kidney. Patients have brain disorders such as a small cerebellum and the absence of specific brain structures.
Microphthalmia with linear skin defects
Microphthalmia with linear skin defects syndrome is a disorder that mainly affects females. In people with this condition, one or both eyes may be very small or poorly developed (microphthalmia). Affected individuals also typically have unusual linear skin markings on the head and neck.
Monosomy 9p syndrome
Monosomy 9p is a rare chromosomal anomaly characterised by psychomotor developmental delay, facial dysmorphism, single umbilical artery, omphalocele, inguinal or umbilical hernia, genital abnormalities, muscular hypotonia, and scoliosis.
Orofaciodigital syndrome
Orofaciodigital syndrome is a group of related conditions that affect the development of the oral cavity (the mouth and teeth), facial features, and digits (fingers and toes).
Panhypopituitarism, X-linked
This syndrome causes a reduced excretion of one or more hormones produced by the pituitary gland, which can lead to dwarfism in children and premature aging in adults. Disorders of gonadotropin secretion affect reproductive functions.
Potocki-Lupski syndrome
Potocki-Lupski syndrome is a rare genetic syndrome caused by duplication on the short arm of chromosome 17. Key features of this syndrome include some degree of developmental delay (primarily cognitive and language deficits), low muscle tone, poor feeding, mild-moderate intellectual delay, and failure to thrive during infancy. In addition, many individuals display some behaviors commonly associated with autism spectrum disorders. Structural cardiovascular anomalies and sleep disturbance have also been frequently reported.
Prader-Willi-like syndrome (SIM1 syndrome)
Prader-Willi-like syndrome is characterised by diminished foetal activity, obesity, muscular hypotonia, intellectual disability, short stature, hypogonadotropic hypogonadism, and small hands and feet.
Rieger syndrome type 1 (RIEG1)
Rieger syndrome is an autosomal dominant disorder of morphogenesis that results in abnormal development of the anterior segment of the eye, and results in blindness from glaucoma in approximately 50% of affected individuals. The syndrome is associate with systemic anomalies including dental hypoplasia, failure of involution of periumbilical skin, and maxillary hypoplasia.
Saethre-Chotzen syndrome (SCS)
Saethre-Chotzing syndrome is characterised by the presence of low-set frontal hairline, an irregular eyelid (ptosis), abnormal ear length, an enlarged parietal foramina (an opening in the cranial skull), a skin formation between fingers, an enlarged thumb, deformation of the fingers, and increased pressure in the brain.
Sensorineural deafness and male infertility
Sensorineural deafness and male infertility is a condition characterised by hearing loss and infertility.
Smith-Magenis syndrome
Smith-Magenis syndrome (SMS) is a rare genetic syndrome caused by a deletion on the short arm of chromosome 17. Although this region contains multiple genes, the loss of one particular gene, RAI1, is responsible for most of the features of the condition. In about 10% of cases, SMS is caused by a genetic change in the RAI1 gene. Key features of this syndrome include mild to moderate intellectual disability, delayed speech and language skills, distinctive facial features, sleep disturbances, and behavioral problems. The prevalence of Smith-Magenis syndrome is 1 in 15,000-25,000.
Split-hand/foot malformation type 3 – Ectrodactyly type 3 (SHFM3)
Ectrodactyly type 3 causes disorders such as fusion of a large number of toes and fingers, split palms and feet, and the absence and/or incomplete development of different bones of the hand, foot, fingers, and toes. Certain patients develop brain development deficiencies, mental disorders, and orofacial clefting.
Split-hand/foot malformation type 5 – Ectrodactyly type 5 (SHFM5)
Split-hand/split-foot malformation (SHFM) is a structural limb malformation which includes deformation of palms and feet, finger jointing, aplasia and/or hypoplasia of the phalanges, metacarpals, and metatarsals. Certain patients develop brain development deficiencies, mental disorders, and orofacial clefting.
Syndrome diaphragmatic hernia, congenital (HCD/DIH)
Congenital diaphragmatic hernia (CDH) refers to a group of congenital defects in the structural integrity of the diaphragm, which is often associated with lethal pulmonary hypoplasia and pulmonary hypertension. Prevalence in newborns ranges from 1 in 2,500 to 1 in 4,000, and there is a 30% to 60% mortality rate.
Trichorhinophalangeal syndrome type I (TRPS1)
Trichorhinophalangeal syndrome type I (TRPS1) is a condition that causes bone and joint malformations, distinctive facial features, and abnormalities of the skin, hair, teeth, sweat glands, and nails. The name of the condition describes some of the areas of the body that are commonly affected: hair (tricho-), nose (rhino-), and fingers and toes (phalangeal).
Van der Woude syndrome (VWS)
Van der Woude syndrome (VWS) is a rare congenital genetic dysmorphic syndrome characterised by paramedian lower-lip fistulae, cleft lip with or without cleft palate, or isolated cleft palate.
11p13 deletion syndrome (WAGR syndrome)
WAGR is an acronym for Wilms tumor, Aniridia, Genitourinary problems, and Range of developmental delays. Key features of this syndrome include complete or partial congenital aniridia (underdevelopment or absence of the iris, which can reduce the sharpness of vision and increase sensitivity to light) and associated eye abnormalities, genitourinary anomalies (such as undescended testicles or hypospadias in males, or internal genital or urinary anomalies in females), variable degrees of intellectual disability and an increased risk of developing Wilms tumors. A minority of patients develop kidney failure, and many patients develop obesity. However, WAGR syndrome with childhood-onset obesity is called WAGRO syndrome.
Wilms tumour 1 (WT1)
Wilms tumour 1 is one of the most common tumours that occur in childhood (1 per 10,000 children) and represents 8% of all cancers in children. It is believed to result from the malignant transformation of abnormally persistent renal stem cells that retain embryonic differentiation potential.
X-linked lymphoproliferative syndrome (XLP)
X-linked lymphoproliferative (XLP) syndrome is an extremely rare inherited immunodeficiency disorder characterised by a defective immune system easily infected with the Epstein-Barr virus, which is the cause of infectious mononucleosis.
Xp11.22–p11.23 microduplication syndrome
Clinical signs of this microdeletion are independent of gender and duplication size. Typical clinical signs are various forms of seizures. Affected individuals are shy and stubborn, and their behaviour is similar to that of a person with autism.
1p36 microdeletion syndrome
1p36 deletion syndrome is characterized by characteristic craniofacial features, intellectual disability, seizures, skeletal abnormalities, and brain and cardiac defects. Prenatal ultrasound may identify certain structural anomalies such as cardiac defects, agenesis of corpus callosum, hydrocephalus, and microcephaly. However, a normal ultrasound does not rule out the underlying anomaly. Lifespan is variable but can be normal.
1q41–q42 microdeletion syndrome
Key features of this syndrome include a significant developmental delay and distinct facial dysmorphisms such as deep-seated eyes, broad nasal tip, and depressed nasal bridge. Additionally, some cases may exhibit conditions such as abnormally small head size (microcephaly), a gap in the roof of the mouth (cleft palate), clubfeet, seizures, and short stature.
2q33.1 deletion syndrome (Glass syndrome)
Key features of the Glass syndrome include an intellectual disability of variable severity and dysmorphic facial features, including an abnormally small lower jaw (micrognathia), downward slanting palpebral fissures, a gap in the roof of the mouth (cleft palate), and crowded teeth.
5q21.1–q31.2 deletion syndrome
The syndrome is primarily recognised based on a significantly modified face, which includes lower forehead, raised eyebrows, a unibrow (synophrys), tight nose, reduced growth of the lower jaw (maxillary prognathism), the enlarged middle part of the upper lip, and narrow lips. The main clinical features are prefrontal and postnatal developmental delay, mental disabilities and abnormal growth of the upper limbs.
8p23.1 deletion syndrome
The clinical manifestations are variable and do not depend on the size of the deletion since it is the same in most patients. Key features of this syndrome include low birth weight, postnatal growth deficiency, mild intellectual deficit, psychomotor retardation, poor speech, seizures, behavioral problems such as hyperactivity and impulsiveness, craniofacial abnormalities (microcephaly, high and narrow forehead, broad nasal bridge, epicanthic folds, high arched palate, short neck, and low set unusually shaped ears), and congenital heart defects.
8p23.1 duplication syndrome
Key features of this syndrome include mild to moderate developmental delay, intellectual disability, mild facial dysmorphism (incl. prominent forehead, arched eyebrows, broad nasal bridge, upturned nares, cleft lip and/or palate) and congenital cardiac anomalies (e.g., atrioventricular septal defect). Other reported features include macrocephaly, behavioral abnormalities (e.g., attention deficit disorder), seizures, hypotonia and ocular and digital anomalies (poly/syndactyly). The prevalence of 8p23.1 duplication syndrome is 1 to 9 in 100,000.
11q11–q13.3 duplication syndrome
The syndrome is characterised by congenital deafness due to an underdeveloped inner ear. The duplication may also cause underdevelopment of the external part of the ears. Very small teeth are also a common feature.
12q14 microdeletion syndrome
Microdeletion 12q14 leads to a mild form of mental disability, early developmental disorders, and osteopoikilosis (excessive bone density leading to pain). Patients are characterised by short stature.
14q11–q22 deletion syndrome
Common features of patients with 14q11–q22 deletion syndrome are microcephaly, hypotonia, poor growth, intellectual disability with poor eye contact, hypoplasia of the corpus callosum and dysmorphic features, including short nose, long philtrum, and flat nasal bridge.
15q26 overgrowth syndrome
Key features of this syndrome include renal anomalies (45%), such as horseshoe kidney and renal agenesis (one or both fetal kidneys fail to develop). Inadequate functioning of the kidneys, backward flow of urine, polycystic kidney and right renal pelvic duplication are common as well. People with this syndrome can also have intellectual disabilities, experience developmental delays, and have a tall stature. Additional features may include craniosynostosis and macrocephaly.
16p11.2–p12.2 microdeletion syndrome
It is accompanied by facial anomalies, including a flattened face, down-slanting palpebral fissures, and eye anomaly. Small ears and mental disability are also typical for this microdeletion.
16p11.2–p12.2 microduplication syndrome
The 16p11.2–p12.2 duplication syndrome is a recurrent genomic disorder with a variable phenotype including developmental delay, dysmorphic features, mild to severe intellectual disability, autism, obsessive or stereotyped behaviour, short stature, and anomalies of the hands and fingers.
17q21.31 deletion syndrome
17q21.31 deletion syndrome or Koolen-de Vries syndrome is a disorder characterised by developmental delay and mild to moderate intellectual disability. People with this disorder are typically described as cheerful, sociable, and cooperative. They usually have weak muscle tone (hypotonia) in childhood. About half have recurrent seizures (epilepsy). Affected individuals often have distinctive facial features, cardiac abnormalities, kidney problems, and skeletal anomalies.
17q21.31 duplication syndrome
The duplication syndrome 17q21.31 refers to short stature, microcephaly, finger anomalies, hirsutism (excessive body hair), atopic dermatitis, digestive problems, facial anomalies (small mouth, anomalies of the ears, small nose) and anomalies of the toes. People with this duplication are mentally challenged, have difficulties in social integration, poor motor skills and behavioural problems (aggression, hyperactivity, obsessive disorders, communication disorders).

Risk of folic acid deficiency during pregnancy
Folic acid is a vitamin B, precisely a water-soluble form of vitamin B9. The human body is not capable of producing it, so we have to consume it with food. Its name comes from the Latin word folium, which means leaf. Coincidence? Not really, because leafy vegetables are an excellent source of folic acid in the diet. Folic acid is found in green vegetables (broccoli, green peppers, lettuce, spinach, Brussels sprouts), peas, lentils, chickpeas, black beans and other legumes, nuts, and certain fruits such as bananas and citrus fruits. However, since the folate found in this type of food is sensitive to high temperatures, light, and oxidation and is also water soluble, avoid prolonged cooking of folate-rich foods.
Folic acid plays a crucial role in the development of the central nervous system in the fetus
Folates play a key role in the synthesis of DNA and amino acids, are involved in cell division, blood production, and immune system function. Folic acid takes care of the formation, metabolism, and proper functioning of the blood cells. It prevents abnormalities in the closing of the neural tube. Therefore, folic acid is particularly important at the beginning of pregnancy, when the neural tube closes, and the embryo’s spine and nervous system begin to form.
Neural tube
The neural tube is a fetal structure from which brains and spinal cord develop. As such, it forms the basis of vital organs and abnormalities in its development cause a group of defects, also called neural tube defects. The process of developing and closing of the neural tube and vertebral arc takes place within the first four weeks after egg fertilisation.
Gynaecologists recommend taking folic acid before a planned pregnancy. Indeed, folic acid consumption during pregnancy has been shown to reduce the chances of abnormal fetal development and neural tube damage by up to 70%.
When a woman finds out she’s pregnant, she is nearing the 5th week of pregnancy. Gynaecologists consider the first day of the last period to be the start of the pregnancy. When a woman performs a test (if she does it immediately), she is already 4 weeks along. Folic acid is crucial during these early weeks; between weeks 8 and 12, it contributes to the development of the fetus and its nervous system. That’s why taking folic acid as a preparation for pregnancy is crucial.

Folic acid levels in women taking the contraceptive pill are alarmingly low
Some studies have shown disorders of vitamin B9 metabolism (folic acid) in women taking oral contraception. After a prolonged period of taking birth control pills, folic acid levels may decrease in the body. However, they are crucial for healthy embryonic development. So when a couple decides to have a child, experts recommend that the woman stops taking birth control pills and the couple uses other methods of protection for three months. During these three months, women should start taking folic acid.
Folic acid is also useful later in pregnancy and during lactation, as it strengthens the immune system and prevents anaemia. Before planned pregnancy, during pregnancy and lactation, gynaecologists recommend 400 micrograms of folic acid daily in the form of dietary supplements.
Higher risk requires higher doses
Couples who have a higher risk of neural tube defects should be much more alert to folic acid needs before and during the planned pregnancy. Usually, in these cases, your doctor also advises a higher dose.
Your risk is higher in the following cases:
- Diabetes
- Epilepsy
- Presence of neural tube defects in the couple planning pregnancy
- Fetus with neural tube defects in past pregnancies
- Family history of neural tube defects
- Consumption of large amounts of alcohol
Frequency of neural tube defects and consequences for the child
If the neural tube does not close properly and completely, the neural tube defects occur.
Neural tube defects are one of the most common defects in babies at birth and affect 300,000 newborns worldwide annually. Among the most common is spina bifida. Depending on the type of neutral tube damage, spina bifida is divided into:
- Spina bifida occulta: one of the most common congenital abnormalities present in a quarter of children in adolescent age. There is a small gap in the spine, but no opening or sac on the back, and it usually doesn’t cause any disabilities. The skin above the deformation can contain a hairy patch, a dimple or a birthmark.
- Meningocele: this type is characterised by a sac of fluid protruding from an opening in the baby’s back. The spinal cord is not in this sac, and there is usually little or no nerve damage.
- Myelomeningocele: the heaviest form of spina bifida. A sack protruding from the baby’s back contains not only fluids but also part of the spinal cord and nerves. Both are damaged and cause moderate to severe disabilities.
